
Can dopamine be depleted from its synapses? (Dopamine 3)
- Hermes Solenzol

- Nov 6, 2025
- 9 min read
Updated: Nov 10, 2025
Dopamine depletion can mean alterations of the synaptic mechanisms that produce, release and eliminate dopamine
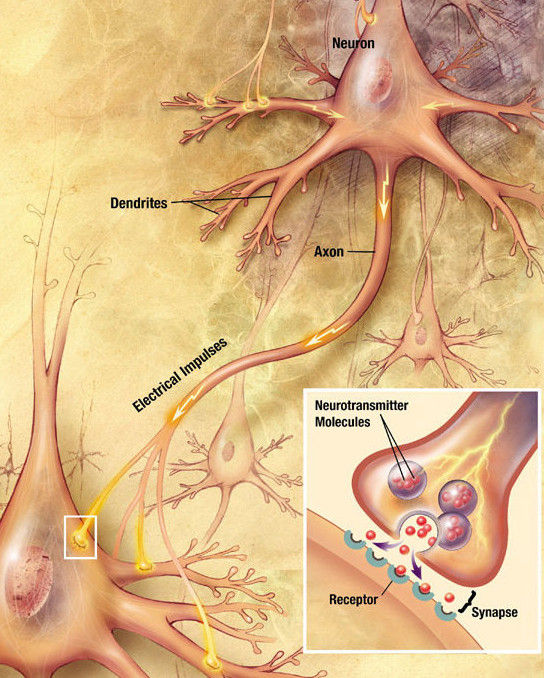
In the two previous articles of this series, I introduced the concept of mental energy and how it is influenced by dopamine. Then I described the reward pathway and the other dopaminergic pathways in the brain.
In this article, I will address the question of whether dopamine can be depleted, because it has been assumed that this leads to a loss of mental energy. To answer that question, we first need to understand the dopamine synapse. That is going to require diving into some heavy-duty molecular neuroscience. So get your dopamine ready and let’s go!
Dopamine neurotransmission - an overview
Neurotransmitters like dopamine operate at the synapse, a tiny junction between the axon of one neuron, the presynaptic neuron, and the dendrites of another, the postsynaptic neuron.
In some synapses, neurons can also make contact with neuronal bodies, axons, muscle cells and endocrine glands. However, the ones that concern us are the ones I describe above, called axodendritic synapses, which are the majority of the synapses in the brain.
In synapses, neurotransmitters like dopamine are stored in synaptic vesicles in the presynaptic terminal.
When electric signals called action potentials reach the presynaptic terminal, the synaptic vesicles fuse with the membrane, releasing dopamine.
Dopamine then crosses the synapse and binds to dopamine receptors, which are proteins in the postsynaptic terminal.
Dopamine receptors send a chemical signal inside the dendrites of the postsynaptic neuron, which determines its firing of action potentials. Some dopamine receptors are located in the presynaptic terminal, where they regulate its function, forming a feedback loop.
The effect of dopamine ends when it gets transported back into the presynaptic terminal by a protein called the dopamine transporter (DAT).
Other transporter, the vesicular monoamine transporter type 2 (VMAT2), puts dopamine back into the synaptic vesicles.
Otherwise, dopamine is destroyed by an enzyme called monoamino oxidase (MAO).
How dopamine is made
Dopamine is made (synthesized) in the brain from two of the 20 amino acids that are strung together to make proteins: L-phenylalanine and L-tyrosine. Don’t worry about the L — it refers to a special configuration of the amino acid molecule. All amino acids in living beings are L (instead of D) stereoisomers.
Dopamine is synthesized through these three reactions catalyzed by enzymes:
L-Phenylalanine → L-Tyrosine → L-DOPA → Dopamine
The first reaction adds a hydroxyl (-OH) group to the benzene ring of phenylalanine, turning it into tyrosine. It is catalyzed by the enzyme phenylalanine hydroxylase.
The second adds yet another -OH group, turning tyrosine into DOPA (dihydroxy-phenylalanine). It is catalyzed by the enzyme tyrosine hydroxylase (TH). This reaction is the limiting step in the production of dopamine, which means that it controls the speed at which it is synthesized.
The last reaction takes away the carboxyl (-COOH) group of DOPA, converting it into dopamine. This is the ‘acid’ group of amino acids, so dopamine is no longer an amino acid, just an amine. Without this group, dopamine has no stereoisomers, that’s why there is no letter L before its name. This reaction is catalyzed by the enzyme DOPA decarboxylase (DDC), also known as aromatic amino acid decarboxylase (AADC).
Dopamine can be converted into norepinephrine and epinephrine (adrenaline) in cells that use them as neurotransmitters (noradrenergic neurons) or as a hormone (cells in the adrenal glands).
What is important here is that tyrosine hydroxylase (TH) is the enzyme that limits the production of dopamine, forming a bottleneck in the series of reactions that make dopamine (Daubner et al., 2011; Bueno-Carrasco et al., 2022). This means that taking L-tyrosine supplements (as advocated by Andrew Huberman in his podcast) will not get you more dopamine in your brain. As we will see, the activity of TH is tightly controlled by signals in the synapse so, no matter how much L-tyrosine you take, these signals will not allow the making of more dopamine. The main signal that inhibits TH activity is one of the dopamine receptors, the D2 receptor.
Too much dopamine causes problems
Unlike L-tyrosine, L-DOPA bypasses the control of TH inhibition. So, if we want to increase dopamine in our brain, we could just take L-DOPA. However, this would be a bad idea.
L-DOPA is used to treat Parkinson’s disease, which is caused by the degeneration of one of the other major dopaminergic pathways: the one that goes from the substantia nigra to the caudate and the putamen nuclei in the dorsal striatum. The death of the dopaminergic neurons in this pathway causes problems with movement, including tremors, rigidity and loss of balance. In its advance stages, Parkinson’s disease leads to sleep problems, mood swings, depression, anxiety and psychosis.
L-DOPA alleviates the symptoms of Parkinson’s disease, particularly in its early stages. Since it bypasses TH, the limiting step in the synthesis of dopamine, L-DOPA supplies the dopamine that is missing from the degenerating neurons.
In fact, taking L-DOPA produces so much dopamine that synaptic vesicles accumulate abnormally large amounts of dopamine, even increasing in size (Sulzer et al., 2016). Then, when each synaptic vesicle fuses with the membrane, it releases abnormally large amounts of dopamine, much more than those induced by natural stimuli.
Another thing that happens in Parkinson’s patients who take L-DOPA is that serotonergic neurons (those that release serotonin) also start synthesizing and releasing dopamine. However, unlike dopaminergic neurons, serotonergic neurons lack ways to eliminate the dopamine that they release, such as the dopamine transporter dopamine or the enzyme monoamine oxidase (MAO) (Riederer et al., 2025). In the advanced stages of Parkinson’s disease, when most dopaminergic neurons in the substantia nigra have died, most of the dopamine produced by L-DOPA comes from serotonergic neurons. This causes a roller-coaster of highs and lows of dopamine, leading to involuntary movements called levodopa-induced dyskinesia.
This is why it’s not a great idea for healthy people to take L-DOPA. We don’t want serotonergic neurons to start making dopamine and messing up our nervous system.
But L-DOPA has some even weirder effects. Some Parkinson’s patients not only regained their sexual function, but started having too many spontaneous erections and nocturnal ejaculations (Bowers et al., 1971; Uitti et al., 1989). Some patients exhibited inappropriate sexual behavior like exhibitionism and excessive use of sex workers (Ceravolo et al., 2010). This hypersexual behavior was also observed in male rats that were given L-DOPA (Tagliamonte et al., 1974). Hypersexuality was more common if the patient was young, male and had early onset Parkinson’s. In other Parkinson’s patients, L-DOPA induces compulsive behaviors like excessive gambling, eating and shopping (Potenza et al., 2007; Ceravolo et al., 2010).
L-DOPA is not the only medication that produces these compulsive behaviors, the selective agonists of dopamine D3 receptors pramipexole and ropinirole cause compulsive behaviors in 30% of the patients who take them (Ahlskog, 2011). The effect of these medications is more pronounced than that of L-DOPA, so dopamine is not what causes the compulsions, but the activation of some of its receptors. Also, it’s not that these patients lose control over their impulses, but that they develop an obsession with the problematic behavior (sex, gambling, shopping, etc.).
There is something odd here. Although these patients developed compulsions to some behaviors, they never got addicted to L-DOPA itself. Addictive drugs are thought to cause addiction by increasing dopamine in the reward pathway. However, L-DOPA increased dopamine without causing addiction. Why?
It seems that something else besides an elevation in dopamine is necessary to trigger addiction. I will try to answer this question when we explore the mechanisms by which drugs cause addiction.

Dopamine release
The mechanisms of neurotransmitter release are quite complex. Each presynaptic terminal contains several types of synaptic vesicles, which contain different neurotransmitters.
Light vesicles contain small molecule neurotransmitters like glutamate, GABA or dopamine.
Dense-core vesicles contain neuropeptides like substance P or endorphins.
Each of these types of vesicle requires a different pattern of incoming action potentials to release its contents: low frequency action potentials for the light vesicles and high frequency for the dense core vesicles (Lever et al., 2001; Adelson et al., 2009).
Although dopamine is released from light synaptic vesicles, there are two different pools of them: one is ready to be released and another that is held in reserve. As the releasable pool of dopamine gets depleted, it is quickly replaced by the reserve pool, which in turn gets refilled with dopamine, either newly synthesized or taken back from the extracellular space by reuptake mechanisms.
Synaptic vesicles are recycled after they release their neurotransmitter (Sulzer et al., 2016). Back inside the presynaptic terminal, they are refilled with dopamine by a protein called vesicular monoamine transporter type 2 (VMAT2).
Dopamine reuptake and degradation
There are membrane proteins, called transporters or uptake mechanisms, whose function is to shuttle molecules across membranes. In particular, the dopamine transporter (DAT) terminates dopamine neurotransmission by taking it out of the extracellular space.
DAT is hugely important because it is responsible for the effect of some addictive drugs. Cocaine produces its effect by inhibiting DAT and thereby increasing the time that dopamine has to activate its receptors. Amphetamines go even further: they reverse the DAT, so instead of putting dopamine back into the cell, it puts even more dopamine out of the cell.
Once inside the cell, dopamine is put back inside the synaptic vesicles by VMAT2. Dopamine is also degraded by a series of enzymes, the most important of which is monoamine oxidase (MAO), which also degrades epinephrine, norepinephrine and serotonin. Some psychoactive medicaments act by inhibiting MAO, thereby increasing the levels of all these neurotransmitters.
What does dopamine depletion actually mean?
In view of all this, the dopamine depletion that is supposed to decrease mental energy can mean several different things.
A decrease in the amount of dopamine stored in the synaptic vesicles. This would be the actual dopamine depletion. It could be caused not only by too much dopamine release but also by a decrease in the synthesis of dopamine or the transport of dopamine back into synaptic vesicles.
Dopamine release could be decreased by things other than its depletion in synaptic vesicles, like less firing of action potential in dopaminergic neurons or alterations in the fusion of synaptic vesicles with the membrane.
Extracellular dopamine could decrease even if the amount of released dopamine stays the same. Thus, the time that dopamine is present outside the neuron to activate dopamine receptors depends on the functioning of the DAT and the enzyme monoamino oxidase. For example, cocaine and amphetamines increase the effect of dopamine by inhibiting or reversing its reuptake by the DAT.
Dopamine receptors could be altered, decreasing their effect even if the amount of extracellular dopamine is the same.
In the next articles, I will examine how these mechanisms are affected by drugs, sex and other pleasurable stimuli to answer the question of whether pleasure decreases mental energy.
As we have seen, the synthesis of dopamine is tightly regulated by the activity of tyrosine hydroxylase, which is controlled by one of the dopamine receptors, the D2 receptor, located in the presynaptic terminal. Thus, any significant decrease in the release of dopamine would mean less activation of these D2 receptors, releasing the inhibition of tyrosine hydroxylase, leading to the synthesis of more dopamine.
The proteins that transport dopamine back into the synaptic vesicles, DAT and VMAT2, quickly take dopamine out from the extracellular space and load it back into the synaptic vesicles. This is shown by the dramatic effect of cocaine and amphetamines, drugs that act by inhibiting DAT.
In fact, the amount of dopamine stored in synaptic vesicles can be increased by taking L-DOPA. This increases compulsive behaviors but, strangely enough, no addition to L-DOPA itself. In the next articles, I will try to explain why.
Therefore, we should discard possibility number 1: that there is any significant depletion of the dopamine stored in synaptic vesicles. In general, high neuronal activity does not lead to the depletion of their neurotransmitters.
A decrease in dopamine release (option number 2) could be caused by less firing of action potentials in the dopaminergic neurons. However, this is a normal part of the functioning of these neurons and it is regulated by what is happening in the rest of the brain. Changes in the fusion of the synaptic vesicles with the membrane can be caused by certain chemicals, but not by physiological mechanisms.
Option number 3 is changes in extracellular dopamine. As I said above, this depends on the activity of DAT, which is very effective unless it is inhibited by cocaine and other psychostimulants.
This leaves the dopamine receptors as the main determinants of the action of dopamine and its possible depletion. We will study them in detail in the next article.
References
Adelson DW, Lao L, Zhang G, Kim W, Marvizón JC (2009) Substance P release and neurokinin 1 receptor activation in the rat spinal cord increases with the firing frequency of C-fibers. Neuroscience 161:538–553.
Ahlskog JE (2011) Pathological behaviors provoked by dopamine agonist therapy of Parkinson's disease. Physiology & behavior 104:168–172.
Bowers MB, Jr., Van Woert M, Davis L (1971) Sexual behavior during L-dopa treatment for Parkinsonism. The American journal of psychiatry 127:1691–1693.
Bueno-Carrasco MT, Cuéllar J, Flydal MI, Santiago C, Kråkenes T-A, Kleppe R, López-Blanco JR, Marcilla M, Teigen K, Alvira S, Chacón P, Martinez A, Valpuesta JM (2022) Structural mechanism for tyrosine hydroxylase inhibition by dopamine and reactivation by Ser40 phosphorylation. Nature Communications 13:74.
Ceravolo R, Frosini D, Rossi C, Bonuccelli U (2010) Spectrum of addictions in Parkinson's disease: from dopamine dysregulation syndrome to impulse control disorders. J Neurol 257:S276–283.
Daubner SC, Le T, Wang S (2011) Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys 508:1–12.
Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M (2001) Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. Journal of Neuroscience 21:4469–4477.
Potenza MN, Voon V, Weintraub D (2007) Drug Insight: impulse control disorders and dopamine therapies in Parkinson's disease. Nat Clin Pract Neurol 3:664–672.
Riederer P, Strobel S, Nagatsu T, Watanabe H, Chen X, Löschmann P-A, Sian-Hulsmann J, Jost WH, Müller T, Dijkstra JM, Monoranu C-M (2025) Levodopa treatment: impacts and mechanisms throughout Parkinson’s disease progression. Journal of Neural Transmission 132:743–779.
Sulzer D, Cragg SJ, Rice ME (2016) Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6:123–148.
Tagliamonte A, Fratta W, Gessa GL (1974) Aphrodisiac effect of L-DOPA and apomorphine in male sexually sluggish rats. Experientia 30:381–382.
Uitti RJ, Tanner CM, Rajput AH, Goetz CG, Klawans HL, Thiessen B (1989) Hypersexuality with antiparkinsonian therapy. Clin Neuropharmacol 12:375–383.
Copyright 2025 Hermes Solenzol.


Comments